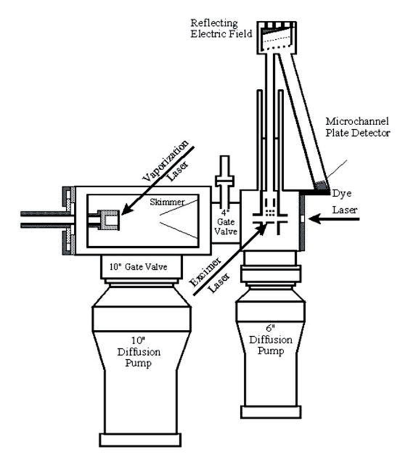
Resonant Two-Photon Ionization (R2PI) Spectrometer
In this instrument, molecules produced in the molecular beam are sent into a time-of-flight (TOF) mass spectrometer that is housed in a high vacuum chamber attached to the source chamber. In the most typical arrangement for this experiment, we direct the output of a tunable laser down the axis of the molecular beam to excite the molecules, and then expose the molecules (20 ns or so later) to the ultraviolet radiation produced by an excimer laser, by the 4th or 5th harmonic of a Nd:YAG laser, or by a frequency-doubled OPO laser. In all cases, the ultraviolet radiation is directed across the axis of the molecular beam. Though there are many other arrangements that can be used in special cases, this is the one that usually works best in R2PI spectroscopy. If we expose the molecules to ultraviolet photons with a short enough wavelength, these photons have enough energy to ionize the molecules in the beam by a one-photon ionization process. This doesn't give us much physical information about the molecule, but it does help us identify which molecules are in the molecule beam. It also allows me an opportunity to describe the TOF mass spectrometer.
If we use a deep ultraviolet laser photon, a large number of ions are produced every time the laser fires. The ions are produced in a static electrostatic field, which accelerates them to a specific kinetic energy. Since the kinetic energy is equal to mv2/2, a correlation between velocity and mass is produced, with lighter ions traveling more rapidly than heavier ones. We detect the ions at the end of a flight tube with a microchannel plate ion detector, and the time between ion production and detection can be converted into mass. By measuring the signal coming out of the detector every 4 ns, we are able to record an entire mass spectrum on each experimental cycle. In this way we can tell what was produced in our source chamber, no matter how complicated the plasma reactions are that lead to the observed products.
Once we have identified what molecules are in the molecular beam, we increase the wavelength of the ultraviolet laser so that no ions are produced by the excimer laser alone. The key to obtaining an optical spectrum is to use photons with an energy that is less than that required to ionize the molecule. The tunable laser pulse is then used to excite the molecule to an excited electronic state. The subsequent ultraviolet laser pulse, with sufficient energy to ionize the excited state but not the ground state, is fired just a few nanoseconds after the first laser. As the first laser is scanned, no ion signal is detected at the mass of interest until a resonant transition is found. When molecules absorb a photon from the first laser beam, exciting them to an excited electronic state, absorption of a photon from the second laser beam then ionizes the molecule. By monitoring the ion signal at a particular mass as the first laser is scanned in wavelength, an absorption spectrum that is specific to that mass may be recorded. Moreover, by simultaneously monitoring the ion signal at several masses, it is possible to record the spectra of several different molecules at the same time. In fact, it is even possible to separately record the spectra of several different isotopic modifications of a given molecule in the same scan.
Given the fact that our source produces a wide variety of chemical species, it is crucial to be able to identify the molecule that is responsible for a given spectroscopic transition. This has proven especially useful in studies we have conducted in which chromium is vaporized in a stream of helium containing 3% CH4. In the same general spectral region, we have been able to identify spectroscopic transitions in CrH, CrC, CrCH, CrCH2, CrCH3, CrC2H, and CrC3H2, all with absolute certainty as to the mass of the absorbing molecule. This is a powerful advantage that the R2PI method enjoys compared to other spectroscopic techniques.
The spectra obtained with the R2PI instrument allow one to locate excited electronic states and measure their vibrational frequencies and anharmonicities. When rotationally resolved spectra are obtained, it is also possible to deduce bond lengths, bond angles, and often the electronic symmetry of the upper and lower states. It is not generally possible to measure the vibrational levels of the ground electronic state, however, since usually only v=0 and possibly v=1 are populated in the supersonic expansion. It is also difficult to probe excited electronic states lying less than 9000 cm-1 above the ground electronic state, because it is difficult to generate photons with tunable frequencies lower than 9000 cm-1. These limitations led us to construct a second instrument, which builds upon the information obtained from the R2PI instrument.