Reaction of Mode-Selected Phenol Cations with Ammonia
1. Reaction of mode-selected phenol cations with ammonia.
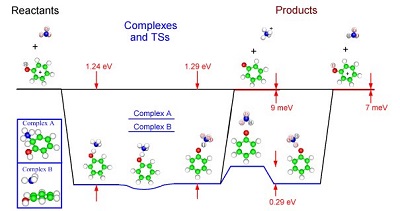 Reactions of C6H5OH+ and C6D5OH+ with ND3 were studied. The energy level scheme for
reaction of PhOH+ with NH3 is shown in Fig. 1. (Zero point-corrected reaction energetics
calculated at G3 level, complexes and transition states at MP2/6-31G*). We have studied
reaction of ground state (GS) ions and ions with one quantum of a 516 cm-1 vibration
(see animation above) and a 815 cm-1 vibration. In the typical benzenoid labeling
scheme, these are assigned as the 6a and 12 modes. Both are in-plane ring modes, and
they differ primarily in that 12 has significant CO stretch character, while in 6a
the COH group moves as a unit. Neither has any significant OH stretch or COH bend
character. We calculate that there are two hydrogen-bonded complexes that have nearly
identical energies. One has the phenol cation hydrogen-bonded to the ammonia nitrogen,
via the hydroxy proton (C6H5OH+ - NH3). In the other, the proton has transferred to
the nitrogen, and the structure is ammonium cation hydrogen-bonded to the oxygen on
a phenoxy radical (C6H5O - NH4+). At the zero point-corrected level, there is no barrier
to interconversion of these complexes, i.e., to intra-complex proton transfer. We
also found complexes corresponding to NH3 complexed to the cloud ("Complex B"), and
NH3 bound to one of the ortho carbon atoms ("Complex A"), however these are substantially
less stable and it is not clear what role they play.
Reactions of C6H5OH+ and C6D5OH+ with ND3 were studied. The energy level scheme for
reaction of PhOH+ with NH3 is shown in Fig. 1. (Zero point-corrected reaction energetics
calculated at G3 level, complexes and transition states at MP2/6-31G*). We have studied
reaction of ground state (GS) ions and ions with one quantum of a 516 cm-1 vibration
(see animation above) and a 815 cm-1 vibration. In the typical benzenoid labeling
scheme, these are assigned as the 6a and 12 modes. Both are in-plane ring modes, and
they differ primarily in that 12 has significant CO stretch character, while in 6a
the COH group moves as a unit. Neither has any significant OH stretch or COH bend
character. We calculate that there are two hydrogen-bonded complexes that have nearly
identical energies. One has the phenol cation hydrogen-bonded to the ammonia nitrogen,
via the hydroxy proton (C6H5OH+ - NH3). In the other, the proton has transferred to
the nitrogen, and the structure is ammonium cation hydrogen-bonded to the oxygen on
a phenoxy radical (C6H5O - NH4+). At the zero point-corrected level, there is no barrier
to interconversion of these complexes, i.e., to intra-complex proton transfer. We
also found complexes corresponding to NH3 complexed to the cloud ("Complex B"), and
NH3 bound to one of the ortho carbon atoms ("Complex A"), however these are substantially
less stable and it is not clear what role they play.
Two product channels are observed. One simply corresponds to H/D exchange, and our labeling experiments show that the exchange occurs only at the hydroxyl site. The energy of this reaction depends on the isotopomer studied, but is in all cases negligible compared to the collision and vibrational energies studied. Nearly isoenergetic is the proton transfer (PT) channel, and again, the energy of the reaction is small compared to the available energy, except for the lowest energies.
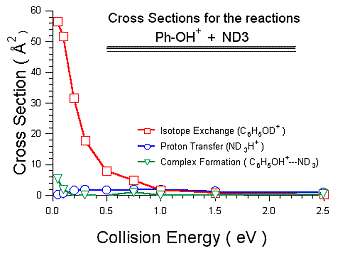 The experimental results are shown in Fig. 2 for reaction of the C6H5OH+ + ND3 isotope
combination, with the cation in its ground vibrational state. One interesting result
is that we actually find a significant effective cross section (i.e., significant
signal) for detecting the [Phenol:ND3]+ collision complex. The implication is that
the lifetime at low collision energies is long enough that a small fraction of the
complexes survive the 1 ms flight time to our detector. It is clear that the lifetime
is in the 100 microsecond range. From the ab initio results to date, it seems likely
that this complex corresponds to the interconverting pair of hydrogen bonded complexes
(C6H5OH+ - NH3 and C6H5O - NH4+). As the collision energy is increased, the complex
still forms but its lifetime rapidly becomes too short to allow direct detection.
The experimental results are shown in Fig. 2 for reaction of the C6H5OH+ + ND3 isotope
combination, with the cation in its ground vibrational state. One interesting result
is that we actually find a significant effective cross section (i.e., significant
signal) for detecting the [Phenol:ND3]+ collision complex. The implication is that
the lifetime at low collision energies is long enough that a small fraction of the
complexes survive the 1 ms flight time to our detector. It is clear that the lifetime
is in the 100 microsecond range. From the ab initio results to date, it seems likely
that this complex corresponds to the interconverting pair of hydrogen bonded complexes
(C6H5OH+ - NH3 and C6H5O - NH4+). As the collision energy is increased, the complex
still forms but its lifetime rapidly becomes too short to allow direct detection.
There is a large cross section for H/D exchange at low collision energies, then the cross section drops rapidly with energy. The corresponding reaction efficiency is about 40% at low energies, dropping to 6% at 1 eV, and becoming negligible at high energies. We propose that this H/D exchange occurs via the pair of hydrogen bonded complexes discussed above. We have also measured the recoil velocity distributions for the H/D exchange product, and find that at all energies, the distribution is forward-backward symmetric, reinforcing the notion that a long-lived complex is required for H/D exchange. As noted, the complex lifetime is quite long at low energies, and the system crosses many times between C6H5OH+ - NH3 and C6H5O - NH4+ configurations. The transition state for the H/D exchange corresponds to the ND3H moiety in the C6H5O - NH4+ complex rotating through a C2v transition state with an activation energy of about 0.29 eV. RRKM calculations indicate that the H/D exchange rate is always much faster than the decomposition rate of the complex, thus for those collisions where the hydrogen-bonded complex forms, the H/D exchange should occur in about three out of four collisions. The fact that the H/D exchange probability is substantially less than 75% and decreases rapidly with collision energy, implies that formation of the hydrogen-bonded complex is not efficient, especially as the collision energy is increased. This may simply reflect the fact that the phenyl group is bulky, and the system may get stuck in a non-reactive collision complex (complex B in the figure).
One real surprise in this system is that there is very little PT (NH4+ production). PT can occur via the same complex-mediated mechanism as H/D exchange (with the complex dissociating from the C6H5O - NH4+ geometry), and also can occur by a direct mechanism. We, therefore, expected PT have a large cross section, greater than or equal to that for H/D exchange. Instead, very little PT is observed and there is a clear threshold (0.18 eV after correction for the collision energy spread). At energies below 2 eV, the PT products have forward-backward symmetric velocity distributions, suggesting that the dominant mechanism is indeed complex-mediated. At high energies, a direct stripping mechanism begins to dominate. The mystery is why the complex formed at low energies almost never breaks up to C6H5O + NH4+. There is no exit channel barrier to NH4+ elimination, and there are no obvious dynamical effects that could suppress the PT channel. We propose that the reaction is actually endoergic. RRKM analysis of the branching ratios indicates an endoergicity of about 160 - 170 meV, which is just at the limit of the expected uncertainties for the G3 energetics and for the previous experimental energetics. Further evidence that the PT channel is suppressed can be inferred from our results on the NH3+ + phenol reaction, not discussed in detail here for space reasons. In NH3+ + phenol, the two most energetically favorable product channels are C6H5OH+ + NH3 (CT) and NH4+ + phenoxy. These are the same products as are observed when starting with C6H5OH+ + ND3 reactants, but in this case both product channels are highly exoergic. As with C6H5OH+ + ND3, very little NH4+ is observed, again suggesting that there is a problem with the existing thermochemistry.
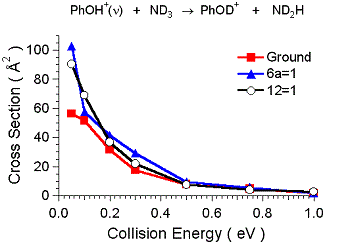 The other surprise was the vibrational effects. For the H/D exchange channel, the
mechanism appears to require a long complex lifetime to allow the exchange. Adding
any energy, including vibration, will reduce the complex lifetime, and might be expected
to give a small inhibition. In fact (see Fig. 4) we see nearly a factor of two enhancement
in H/D exchange from both vibrations, and the effect appears slightly larger for the
lower energy 6a vibration. Because all mode sensitivity will be lost in a long-lived
complex, the implication is that these vibrations enhance the probability of forming
the complex. Indeed, the branching ratio suggests that complex formation is nearly
100% efficient for vibrationally excited reactants at low energies (compared to 50%
for ground state PhOH+. These vibrations are both in-plane ring distortions, and it
is not obvious how these might enhance complex formation. One possibility is that
they enhance "diffusion" of the NH3 moiety in complex B (see figure) enhancing conversion
to the hydrogen bonded complex. This is the first example of such an effect in a molecule
this size.
The other surprise was the vibrational effects. For the H/D exchange channel, the
mechanism appears to require a long complex lifetime to allow the exchange. Adding
any energy, including vibration, will reduce the complex lifetime, and might be expected
to give a small inhibition. In fact (see Fig. 4) we see nearly a factor of two enhancement
in H/D exchange from both vibrations, and the effect appears slightly larger for the
lower energy 6a vibration. Because all mode sensitivity will be lost in a long-lived
complex, the implication is that these vibrations enhance the probability of forming
the complex. Indeed, the branching ratio suggests that complex formation is nearly
100% efficient for vibrationally excited reactants at low energies (compared to 50%
for ground state PhOH+. These vibrations are both in-plane ring distortions, and it
is not obvious how these might enhance complex formation. One possibility is that
they enhance "diffusion" of the NH3 moiety in complex B (see figure) enhancing conversion
to the hydrogen bonded complex. This is the first example of such an effect in a molecule
this size.
2. Rotational effects in NO+ + EtOH.
In this study, we probed the effects of reactant rotation and collision energy on the hydride abstraction reaction:
NO+ (N+ = 0 or 10) + C2H5OH -> HNO + C2H5O+
The results are shown in Fig. 5. This reaction is exoergic with no barrier, and the thermal rate constant indicates about 50% reaction efficiency under thermal conditions. This is rather typical of such reactions, and is usually taken to imply a capture-controlled reaction mechanism.
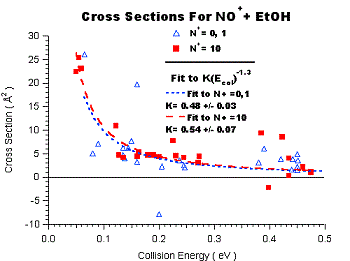 We find that the reaction actually has an extremely sharp collision energy dependence.
(This sharp dependence is why the data appear so noisy. Because there are always small
run-to-run changes in the ion kinetic energy, we can't average the points together).
The cross section is found to vary as 1/Ecol1.3 in the collision energy range above
50 meV, compared to the 1/E0.5 behavior expected for a capture-controlled reaction.
At 50 meV, the reaction efficiency is already only 9%, and by 1 eV, it is only 1 %.
Such sharp Ecol dependence is often interpreted as the sign of a severe phase-space
bottleneck to reaction, the idea being that at very low energies reaction is mediated
by a weakly-bound complex that provides time for the reactants to find the correct
configuration leading to reaction. As the collision energy is raised, the lifetime
of this complex drops rapidly, thus reducing the reaction efficiency.
We find that the reaction actually has an extremely sharp collision energy dependence.
(This sharp dependence is why the data appear so noisy. Because there are always small
run-to-run changes in the ion kinetic energy, we can't average the points together).
The cross section is found to vary as 1/Ecol1.3 in the collision energy range above
50 meV, compared to the 1/E0.5 behavior expected for a capture-controlled reaction.
At 50 meV, the reaction efficiency is already only 9%, and by 1 eV, it is only 1 %.
Such sharp Ecol dependence is often interpreted as the sign of a severe phase-space
bottleneck to reaction, the idea being that at very low energies reaction is mediated
by a weakly-bound complex that provides time for the reactants to find the correct
configuration leading to reaction. As the collision energy is raised, the lifetime
of this complex drops rapidly, thus reducing the reaction efficiency.
Curiously, although collision energy has a very strong inhibiting effect on the reaction, NO+ rotational excitation (N+ = 10 = 200 cm-1) has little if any effect. At our lowest collision energy, this much rotational excitation would cause a nearly 50% drop in cross section if rotational and collision energy were equivalent. Instead, there is a small increase, but not outside the experimental uncertainty. The very few previous studies of rotational effects in ion-molecule reactions have found effects of rotation to be comparable to those of collision energy.
We are currently working on ab initio calculations to attempt to find the origin of the very strong collision energy dependence and negligible rotational effect. We have located a number of possible intermediate complexes and are examining transition states that may control the reaction.