Richard D. Ernst
 INORGANIC CHEMISTRY
INORGANIC CHEMISTRY
Professor
B.S., University of California, Berkeley, 1973
Ph.D., Northwestern University, 1977
Phone: (801) 581-8639
Office: 2166 HEB-N
Email: ernst@chem.utah.edu
Richard is not recruiting new Ph.D. graduate students into his lab. Undergraduates
are welcome to apply for research positions in his lab.
Activities & Awards
- NSF Creativity Award, 1987-89
- AAAS Fellow, 1991
- ASUU Student Choice Awards for Excellence in Teaching, 1997 and 2002
- University of Utah Distinguished Teaching Award, 2003
- Robert W. Parry Teaching Award, 2003
Research Interests
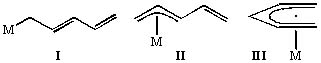
Our most intensive efforts have been applied to the synthesis, reaction studies and structural characterization of transition metal complexes containing pentadienyl ligands. These ligands seem interesting as they can be considered to be “opened” cyclopentadienyl ligands. One might therefore expect that a large area of chemistry would exist wherein a pentadienyl ligand replaces its more familiar cyclic counterpart (C5H5). A likely advantage of such a ligand should be a greater tendency to exist in monohapto (I) and trihapto (II) bonding configurations as well as pentahapto (III). Isomerizations between these forms can be very beneficial chemically as well as catalytically, as a mechanism then exists for either bringing on additional ligands or expelling others. For these reasons one can expect metal pentadienyl complexes to be chemically very reactive. Further, since η5-bound pentadienyl is in essence a better chelate than allyl, a wide variety of metal-pentadienyl complexes should be thermally stable. Thus, a major goal of our work in this area has been to investigate both the physical and chemical natures of these systems and thereby gain a more unified understanding of these ligands.
The potential of these ligands can be appreciated from the complex Ti(C5H5)(2,4-C7H11)(PEt3) (C5H5 = cyclopentadienyl; C7H11 = dimethylpentadienyl). A structural study has revealed the Ti-C bonds for the open pentadienyl ligand to be much shorter than those for the C5H5 ligand, ca. 2.24 vs. 2.35 Å. Nevertheless, this compound reacts readily with a wide variety of unsaturated organic molecules (e.g., imines, ketones, alkynes, nitriles, and isonitriles), leading to selective coupling between the unsaturated molecule and the open pentadienyl ligand. Thus not only is pentadienyl more strongly bound than cyclopentadienyl, it is also more reactive. In fact, a number of applications of these compounds have been developed, and some open metallocenes of iron and ruthenium are sold commercially.
The coupling reactions of the above half-open titanocene with alkynes have led to particularly notable results. In some of the products, rearrangements occur via C–C bond cleavage reactions, while other products actually displayed agostic (C–C)→Ti interactions, and were the first such species to be isolated. Confirmation of the presence of such interactions was provided by the observation of lengthened C–C bonds, remarkably low C(13)–C(13) coupling constants, theoretical studies, and a careful bonding electron density study. Ultimately it was recognized that C–C agostic interactions are present in electron deficient metallacyclobutanes and metallacyclobutenes, and play a role in both olefin polymerization and metathesis reactions.
Due to pentadienyl ligands’ generally strong δ acidities, and large girths, they have displayed a strong preference for bonding to metals in +2 or lower oxidation states. However, in recent years some high valent species have been isolated, e.g., Zr(C5H5)(6,6-dmch)X2 (dmch = dimethylcyclohexadienyl; X = Cl, Br, I). Structural data for these species nicely correlate with expectations, as the contracted Zr(IV) orbitals are not able to interact effectively with all five delocalized carbon atoms of the 6,6-dmch ligand, leading to a short Zr–C interaction for the 3 position, and progressively longer Zr–C bonds on going to the 6,6-dmch termini. Thus, the 6,6-dmch ligand is showing a pronounced slippage from the Zr(IV) coordination sphere. In addition, while the low valent complexes show shorter M–C distances for the electronically open dienyl ligands (vide supra), just the opposite is observed for the Zr(C5H5)(6,6-dmch)X2 complexes, whose Zr–C (C5H5) distances are now ca. 0.1 Å shorter than those for 6,6-dmch, reflecting a dramatic reversal in bonding favorabilities.
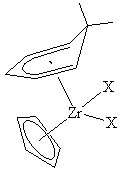
In efforts to prepare higher valent pentadienyl complexes containing fluoride ligands, we began to look at possible reactions of SF6 with low valent metal compounds. SF6 had been regarded as extraordinarily inert, having an average lifetime of over 3000 years in the atmosphere, which contributes to its having the highest global warming potential of any known gas. Despite SF6’s legendary inertness, there seemed to be no good reason why electropositive, low valent metals shouldn’t be able to abstract fluorines from SF6. Indeed, we found that some reactions involving SF6 could take place even at low temperatures, and SF6 could even be more reactive than some reactive fluorinating agents such as XeF2 or CoF3.
Selected Publications
- R. Tomaszewski, I. Hyla-Kryspin, C.L. Mayne, A.M. Arif, R. Gleiter, and R.D. Ernst, “Shorter Nonbonded Than Bonded Contacts or Nonclassical Metal-to-Saturated Carbon Atom Interactions?,” J. Am. Chem. Soc. 120, 2959-2960 (1998).
- S. Pillet, G. Wu, V. Kulsomphob, B.G. Harvey, R.D. Ernst, and P. Coppens, “Investigation of Zr-C, Zr-N, and Potential Agostic Interactions in an Organozirconium Complex by Experimental Electron Density Analysis,” J. Am. Chem. Soc. 125, 1937 (2003).
- A. Rajapakshe, N.E. Gruhn, D.L. Lichtenberger, R. Basta, A.M. Arif, and R.D. Ernst, “Pentadienyls vs. Cyclopentadienyls and Reversal of Bonding Affinity with Metal Oxidation State: Synthesis, Molecular Structures, and Electronic Structures of High-Valent Zirconium Pentadienyl Complexes,” J. Am. Chem. Soc. 126, 14105-14116 (2004).
- R. Basta, B.G. Harvey, A.M. Arif, and R.D. Ernst, “Reactions of SF6 with Organotitanium and Organozirconium Complexes: The ‘Inert’ SF6 as a Reactive Fluorinating Agent,” J. Am. Chem. Soc. 127, 11924-11925 (2005).
- B.G. Harvey, C.L. Mayne, A.M. Arif, R. Tomaszewski, and R.D. Ernst, “Structural and Spectroscopic Demonstration of Agostic C–C Interactions in Electron-Deficient Metallacyclobutanes and Related Cage Complexes: Possible Implications for Olefin Polymerizations and Metatheses,” J. Am. Chem. Soc. 127, 16426-16435 (2005) [corrn.: 128, 1770 (2006)].
- B.G. Harvey, A.M. Arif, and R.D. Ernst, “Coupling Reactions of Alkynes With Half-Open Titanocenes: Agostic (C–C)→Ti Interactions in a Tetra(alkyne) Coupling Product with the Ti(C5H5)(c-C8H11) Fragment,” J. Organometal. Chem. 691, 5211-5217 (2006).
- B.G. Harvey, A.M. Arif, A. Glöckner, and R.D. Ernst, “SF6 as a Selective and Reactive Fluorinating Agent for Low-Valent Transition Metal Complexes,” Organometallics 26, 2872-2879 (2007).
- L. Stahl and R.D. Ernst, “Pentadienyl Chemistry of the Group IV Transition Metals,” Advances in Organometallic Chem. 55, 137-199 (2008).
- S. Scheins, M. Messerschmidt, M. Gembicky, M. Pitak, A. Volkov, P. Coppens, B.G. Harvey, G.C. Turpin, A.M. Arif, and R.D. Ernst, “Charge Density Analysis of the (C–C)→Ti Agostic Interactions in a Titanacyclobutane Complex,” J. Am. Chem. Soc. 131, 6154-6160 (2009).
-
A. Glöckner, M. Tamm, A.M. Arif, and R.D. Ernst, “A New Versatile Approach to Substituted Cyclopentadienyl-Cycloheptatrienyl Complexes of Zirconium (Trozircenes),”Organometallics 28, 7041-7046 (2009).
-
Y. Shi, A.M. Arif, and R.D.Ernst, "Use of Pyrazolyl Ligands for the Formation of a Bimetallic Cobalt-Ruthenium Complex," Polyhedron, 30, 1899-1905 (2011).
-
S.Bali, M.A. Tofanelli, R.D. Ernst and E.M. Eyring, “Chromium(III) catalysts in ionic liquids for the conversion of glucose to 5-(hydroxymethyl)furfural (HMF): Insight into metal catalyst:ionic liquid mediated conversion of cellulosic biomass to biofuels and chemicals,” Biomass and Bioenergy 42, 224-227 (2012).
-
B.G. Harvey and R.D. Ernst, “Transition-Metal Complexes with (C-C)àM Agostic Interactions,” European Journal of Inorganic Chemistry, 1205-1226 (2017).