Simulations enable “choose-your-own-adventure” stereochemistry
by Paul Gabrielsen
Stereochemistry is a science of reflection. Two chemical molecules with the same composition and structure, but with one as the mirror image of the other, can produce wildly varying effects. Controlling which molecule emerges from a given reaction is a critical, but sometimes poorly understood, process.
But University of Utah chemist Matt Sigman has been developing a way to get a better grasp on this tricky field of chemistry. He’s been using tools formerly in the realm of the pharmaceutical industry to predict, using computer models, how certain molecules will behave in certain reactions. He’s already applied this method to battery chemistry and now he and colleagues at the City University of New York are using it to take some of the mystery out of stereochemistry. Their work will be published in Science on September 20, 2018.
Their focus is on the Suzuki cross-coupling reaction, a Nobel Prize-winning reaction that forges a carbon-carbon bond in a molecule. It’s often used to synthesize biaryls, common motifs in pharmaceutical molecules. Extending this powerful transformation to the synthesis of alkyl arenes requires the use of alkylboron starting materials.
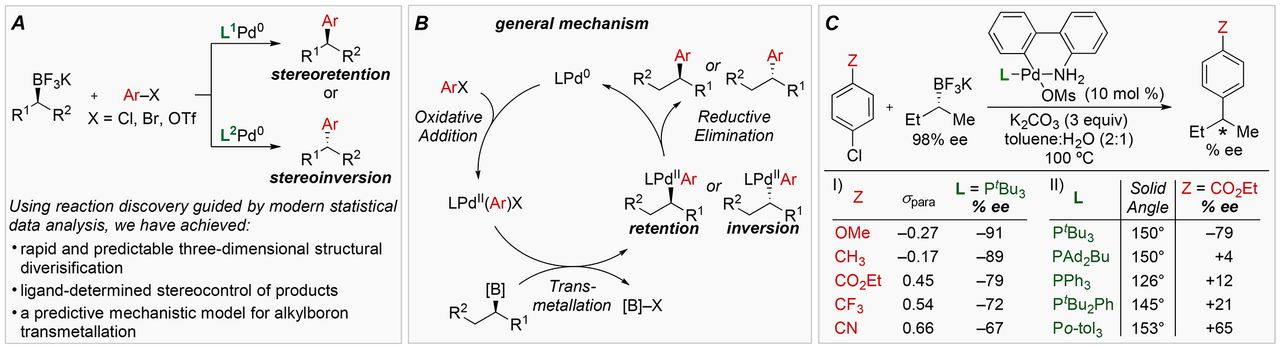
Fig. 1 Reaction development.
(A) Enantiodivergent Suzuki reactions of secondary alkylboron nucleophiles. (B) General mechanism. (C) Initial investigation of substrate and ligand influences on stereoselectivity. +% ee = net retention, –% ee = net inversion.
Here’s where stereochemistry comes in: these starting materials can produce two different products with two different structures, and it’s not well known what conditions lead to one structure or the other.
And here’s where Sigman and his colleagues come in: Their simulations of phosphine molecules (ligands, once they’re attached to a catalyst) generated predictive statistical models of how the phosphine would affect the reaction.
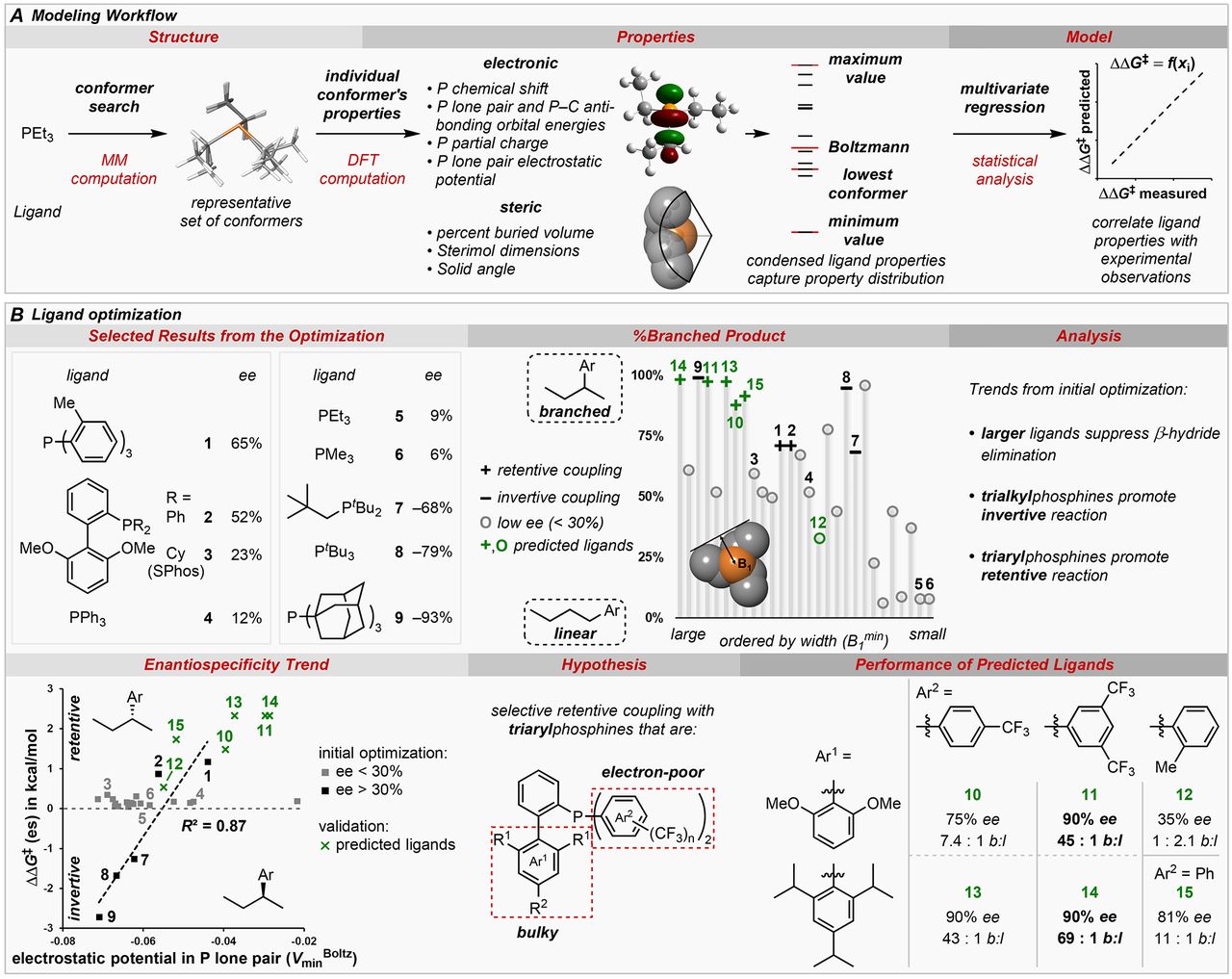
Fig. 2 Phosphine parameterization.
(A) Workflow of parameter generation and statistical modeling. (B) Application of phosphine parameterization to ligand optimization of the reaction shown in Fig. 1C with Z = CO2Et. b:l = branched to linear ratio. +% ee = net retention, –% ee = net inversion.
“We used our data-driven tools to derive significant insight into how the process works that allows us to design the correct additives to get the desired outcomes,” Sigman said. The results allow chemists to control which stereochemical product comes out of the reaction, simply by selecting the right ligand. It’s more than just a laboratory convenience, though. The study also reveals much more about how this important chemical process works.
More information, including a copy of the paper, can be found online at the Science press package at http://www.eurekalert.org/jrnls/sci. You will need your user ID and password to access this information.
The full study can be found at http://science.sciencemag.org/lookup/doi/10.1126/science.aat2299
Contacts:
--Matt Sigman, distinguished professor, Department of Chemistry, 801-585-0774, sigman@chem.utah.edu
--Paul Gabrielsen, science writer, University of Utah Communications, 801-585-6861, paul.gabrielsen@utah.edu